Ciencia
Hacia nuevos tratamientos antivirales
BUSCAN FRENAR AL DENGUE CON SIMULACIONES POR COMPUTADORA
jueves 6 de agosto de 2026
Mediante supercomputadoras, una científica de la UBA busca identificar los puntos débiles del dengue para abrir el camino a nuevos tratamientos antivirales de amplio espectro. Su trabajo fue reconocido con el premio L’Oréal-UNESCO “Por las Mujeres en la Ciencia” 2025.

Mehrnoosh Arrar, investigadora del Instituto de Cálculo de la Facultad de Ciencias Exactas y Naturales de la UBA.
A través de simulaciones computacionales, un estudio de la Universidad de Buenos Aires busca identificar los puntos más vulnerables en la estructura del virus del dengue para identificar los sitios donde una futura droga podría detener la infección antes de que se propague.
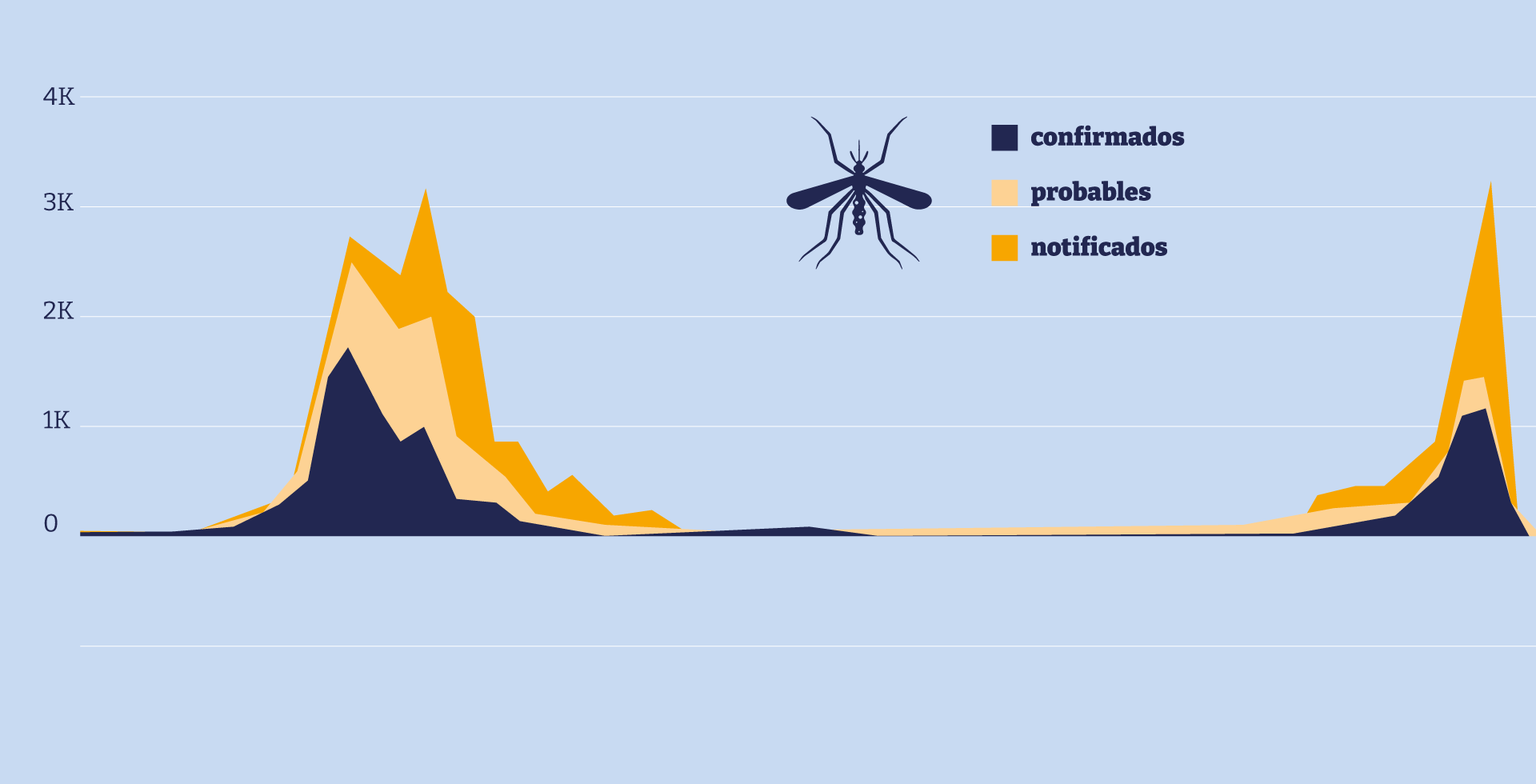
Cada año, cientos de millones de personas contraen dengue en el mundo. La mayoría se recupera, pero una parte desarrolla una forma severa que puede ser letal. Sin embargo, no existe ningún antiviral que frene la replicación del virus: los médicos solo pueden tratar los síntomas y esperar.
Mehrnoosh Arrar, investigadora del Instituto de Cálculo de la Facultad de Ciencias Exactas y Naturales de la UBA, trabaja para cambiar eso, aunque no desde un laboratorio con tubos de ensayo, sino desde una computadora.
Arrar recibió la categoría Beca del Premio Nacional L'Oréal-UNESCO "Por las Mujeres en la Ciencia" 2025 por su proyecto "Diseño racional de antivirales para el Dengue". El galardón, que en su decimonovena edición argentina reconoció a seis científicas del país, le otorgó diez millones de pesos para continuar su trabajo.
"En el contexto actual de desfinanciamiento brutal de la ciencia argentina, este premio representa mucho", sostuvo Arrar. "Es un reconocimiento a un esfuerzo colectivo de quienes seguimos buscando maneras de continuar haciendo ciencia de calidad desde Argentina, a pesar de un contexto alarmante. Y el financiamiento otorgado nos da una posibilidad de seguir avanzando con este proyecto."

El motor molecular que le interesa al virus
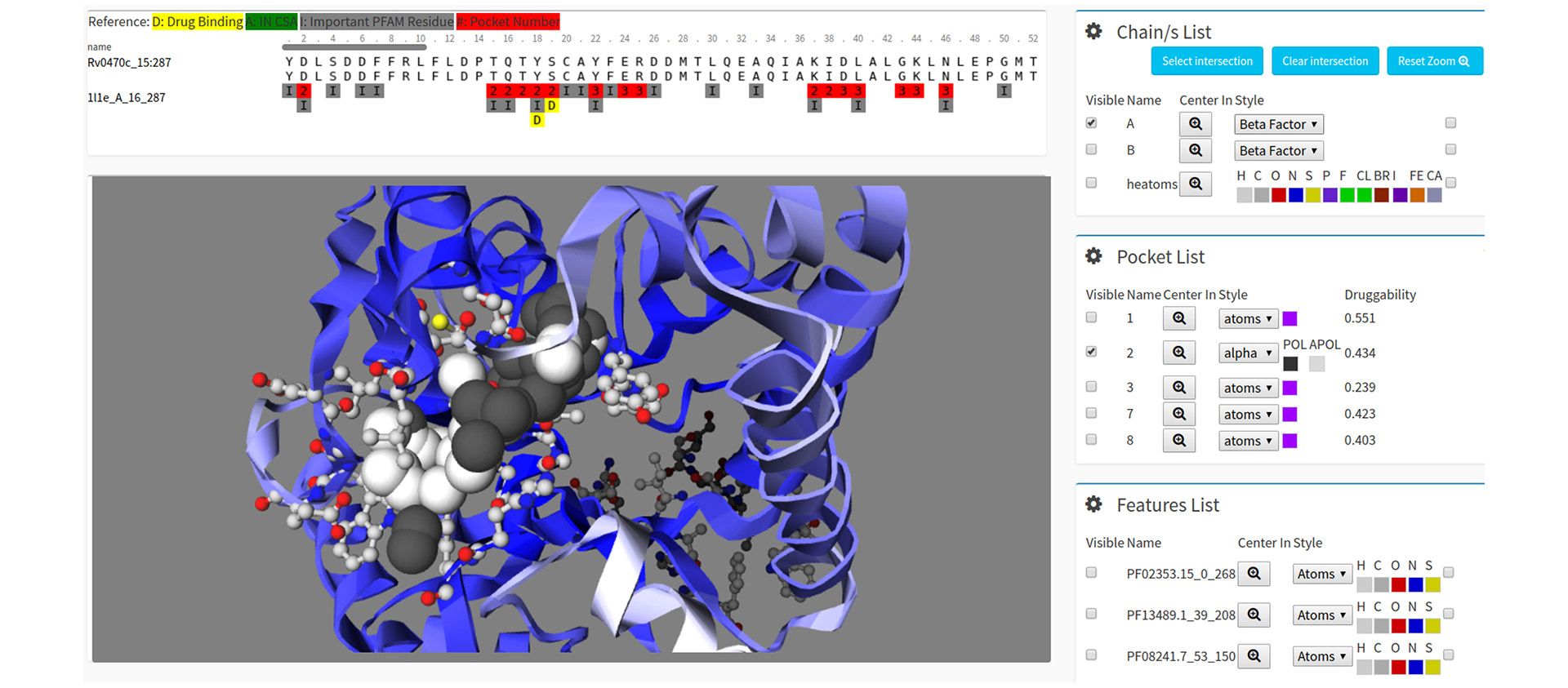
El corazón de la investigación se centra en comprender el funcionamiento de una proteína específica del virus, llamada helicasa. Para entenderlo de forma sencilla, esta estructura actúa como un pequeño motor molecular. Con el fin de ponerse en marcha, consume unas moléculas llamadas ATP, que funcionan como el combustible de un automóvil.
"La helicasa tiene un rol indispensable para la replicación viral, y eso es lo que ha sido el foco de nuestra investigación", explicó Arrar, investigadora del Instituto de Cálculo de la Facultad de Ciencias Exactas y Naturales de la UBA. “El objetivo de largo plazo es encontrar moléculas que interfieran en ese funcionamiento y, en consecuencia, impidan que el virus se multiplique”.
Para estudiar estos procesos, Arrar y su equipo recurren a una herramienta informática llamada dinámica molecular. A partir de estructuras tridimensionales obtenidas experimentalmente, que muestran la posición de cada átomo de una proteína, emplean supercomputadoras para simular cómo esos átomos interactúan y se mueven a lo largo del tiempo siguiendo las leyes de la física
"La técnica calcula el movimiento de miles de átomos mediante miles de millones de pasos extremadamente pequeños, reproduciendo en la computadora el comportamiento dinámico de las biomoléculas”, explicó Arrar. “El resultado no es una fotografía estática, sino una trayectoria a escala atómica que nos permite observar cómo cambia la proteína, cómo interactúa con otras moléculas y qué mecanismos hacen posibles su función biológica”.
El objetivo central del proyecto consiste en encontrar cómo romper la maquinaria de la que se vale el virus para replicarse. Actualmente, el equipo trabaja en identificar y caracterizar las llamadas redes alostéricas, es decir, entender cómo se comunican distintos sitios de las biomoléculas, y qué pasa cuando se "interrumpe" esa comunicación.
Diálogo constante con el laboratorio
Las simulaciones de dinámica molecular permiten generar hipótesis sobre el comportamiento de las biomoléculas, pero esas predicciones deben contrastarse con evidencia experimental. Por eso, la investigación de Arrar existe en permanente conversación con grupos experimentales que pueden validar, o refutar, sus predicciones en situaciones reales.
Por este motivo, el equipo de Arrar trabaja en estrecha colaboración con el grupo de Sergio Benjamín Kaufman, investigador en la Facultad de Farmacia y Bioquímica de la UBA, quien aporta datos precisos sobre la cinética y velocidad molecular de la proteína para refinar los códigos informáticos. Asimismo, cooperan con el equipo de Andrea Gamarnik, en el Instituto Leloir, quienes se encargan de evaluar las hipótesis directamente en vivo, analizando, por ejemplo, cómo impactan las moléculas seleccionadas en la replicación real del virus dentro de las células.
"Podemos proponer una hipótesis a partir de las simulaciones, y nos pueden decir si tiene sentido a la luz de los datos observados experimentalmente. Cuando aparecen discrepancias, eso nos obliga a repensar cómo estamos modelando el sistema", reflexionó Arrar. El resultado es un ciclo de refinamiento continuo donde la simulación y el experimento se retroalimentan.
Así fue que recientemente, este equipo multidisciplinario, logró avances importantes. Descubrieron un mecanismo clave que al ser interferido con un compuesto químico, se frenaba la replicación viral. Si bien es un resultado esperanzador, publicado en la revista especializada Plos Pathogens, todavía faltan muchos estudios y controles antes de llegar a una aplicación.
Más allá del dengue
El conocimiento generado por este tipo de investigación tiene aplicaciones que van más lejos que el dengue. Los flavivirus, familia que incluye también al zika y al fiebre amarilla, comparten mecanismos de replicación similares. Si la helicasa del dengue revela sus puntos débiles, hay buenas posibilidades de que esa información sea transferible a sus parientes.
"Me parece interesante pensar en no solo el virus del dengue, sino también en otros virus emergentes o reemergentes que podrían estar utilizando mecanismos similares. Este conocimiento básico es relevante para el dengue, pero también puede ayudarnos a estar mejor preparados frente a problemas que quizás hoy no son prioritarios, pero que podrían serlo en el futuro”, señaló Arrar.
Incluso el desarrollo de mejores vacunas podría beneficiarse. Las vacunas utilizan versiones del virus atenuadas que han sido modificadas para que no se repliquen eficientemente. Entender en detalle cómo funciona la helicasa abre la posibilidad de diseñar nuevas mutaciones que debiliten esa capacidad de replicación de manera controlada.
Los tiempos de la ciencia
Aunque las simulaciones aceleran notablemente la búsqueda al filtrar miles de candidatos químicos de forma virtual, el camino hacia un medicamento real requiere tiempo y varias etapas. Una vez seleccionadas las moléculas ideales por computadora, el proceso continuará con ensayos en el laboratorio y, eventualmente, requerirá el diseño de alianzas estratégicas entre el sector público y la industria farmacéutica privada para financiar su desarrollo a gran escala.
Si bien es un desafío de largo aliento, el entendimiento de la ciencia básica ya ha comenzado a trazar el mapa. Y el uso de la informática para descifrar la biología molecular vive una auténtica revolución global.
Esta metodología ya había sido respaldada internacionalmente en 2013, cuando el Premio Nobel de Química reconoció a los desarrolladores de métodos para simular sistemas químicos complejos, y más recientemente con los revolucionarios avances en inteligencia artificial aplicados al diseño y predicción de estructuras de proteínas.
En esa misma línea de vanguardia, el proyecto de la UBA demuestra cómo el modelado predictivo se ha vuelto una herramienta estratégica esencial para anticipar soluciones frente a los desafíos epidemiológicos del siglo XXI.